
一、✌ 聚类算法
1.1 ✌ 无监督学习与聚类算法
| 聚类 | 分类 | |
| 区别 | 不经过学习,将样本数据分为多个簇 | 将样本数据导入已经学习过的模型进行学习,有标签可以进行参考 |
| 具体算法 | DBSCAN、KMeans、层次聚类等 | 逻辑回归、随机森林、决策树、贝叶斯等 |
| 算法评估 | 一般来说,聚类结果是不确定的,只是根据特征进行分类,没有进行监督学习 | 分类结果是唯一的 |
| 分类:根据已经建好的模型或者数据,将新的数据按照相关算法进行分类处理 | ||
| 聚类:将原样本数据直接进行相关算法进行分类 |
1.2 ✌ 聚类常用算法
1.2.1 ✌ KMeans算法的基本原理
KMeans中的K代表簇的数量,Means代表每个簇中的均值,就是各个分类的中心点,KMeans算法以计算样本间距离为样本相似度的度量标准,将距离相似的样本分到一个簇中。
样本间距离计算方式有:欧式距离、余弦相似度、曼哈顿距离,一般来说采用欧式距离,对于文本可能会使用余弦相似度。
KMeans算法核心:
- 随机从样本中选定k个样本点
- 根据欧式距离度量每个样本对于样本点的距离,将每个样本分到距离最近的样本点的簇中
- 根据新的簇的分类结果,再从每个簇中选取新的样本点,这次的样本点为每个簇中的中心点
- 不断循环2、3步骤,知道每个簇中的样本中心不变
2.2 ✌ 重要参数n_clusters
这个参数就为KMeans中的k,即所要分的类的数量,我们用一串代码来测试这个参数
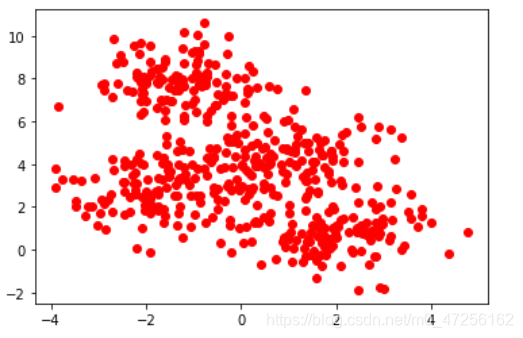
import pandas as pd from sklearn.datasets import make_blobs x,y=make_blobs(n_samples=500,n_features=2,centers=4,random_state=123)
第三行的代码是为了产生自定义数据集,参数分别为样本数量,样本特征,中心,和随机种子
fig,ax1=plt.subplots(1) ax1.scatter(data[0],data[1],c='red') plt.show()
#采用KMeans from sklearn.cluster import KMeans kms=KMeans(n_clusters=4) kms.fit(x) label_kms=kms.labels_ color=['red','green','yellow','blue'] plt.figure(figsize=(6,6)) for i in range(4): plt.scatter(x[label_kms==i]['特征1'],data[label_kms==i]['特征2'],c=color[i]) plt.show()
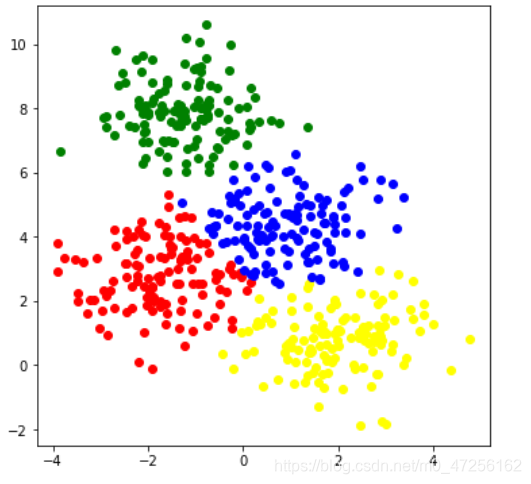
首先初始化模型类
然后训练模型
labels_,该属性为样本的分类值
#采用KMeans from sklearn.cluster import KMeans kms=KMeans(n_clusters=4) kms.fit(x) label_kms=kms.labels_ color=['red','green','yellow','blue'] plt.figure(figsize=(6,6)) for i in range(4): plt.scatter(x[label_kms==i]['特征1'],data[label_kms==i]['特征2'],c=color[i]) plt.show()
2.3 ✌ 重要参数init & random_state & n_init
在K-Means中有一个重要的环节,就是放置初始质心。如果有足够的时间,K-means一定会收敛,但Inertia可能收
敛到局部最小值。是否能够收敛到真正的最小值很大程度上取决于质心的初始化。init就是用来帮助我们决定初始
化方式的参数。
初始质心放置的位置不同,聚类的结果很可能也会不一样,一个好的质心选择可以让K-Means避免更多的计算,让
算法收敛稳定且更快。在之前讲解初始质心的放置时,我们是使用”随机“的方法在样本点中抽取k个样本作为初始质
心,这种方法显然不符合”稳定且更快“的需求。为此,我们可以使用random_state参数来控制每次生成的初始质心
都在相同位置,甚至可以画学习曲线来确定最优的random_state是哪个整数。
一个random_state对应一个质心随机初始化的随机数种子。如果不指定随机数种子,则sklearn中的K-means并不
会只选择一个随机模式扔出结果,而会在每个随机数种子下运行多次,并使用结果最好的一个随机数种子来作为初
始质心。我们可以使用参数n_init来选择,每个随机数种子下运行的次数。这个参数不常用到,默认10次,如果我
们希望运行的结果更加精确,那我们可以增加这个参数n_init的值来增加每个随机数种子下运行的次数。
然而这种方法依然是基于随机性的。
为了优化选择初始质心的方法,2007年Arthur, David, and Sergei Vassilvitskii三人发表了论文“k-means++: The
advantages of careful seeding”,他们开发了”k-means ++“初始化方案,使得初始质心(通常)彼此远离,以此来
引导出比随机初始化更可靠的结果。在sklearn中,我们使用参数init ='k-means ++'来选择使用k-means ++作为质
心初始化的方案。通常来说,我建议保留默认的"k-means++“的方法。
init:可输入"k-means++”,“random"或者一个n维数组。这是初始化质心的方法,默认"k-means++”。输入"kmeans++":一种为K均值聚类选择初始聚类中心的聪明的办法,以加速收敛。如果输入了n维数组,数组的形状应
该是(n_clusters,n_features)并给出初始质心。
random_state:控制每次质心随机初始化的随机数种子
n_init:整数,默认10,使用不同的质心随机初始化的种子来运行k-means算法的次数。最终结果会是基于Inertia
来计算的n_init次连续运行后的最佳输出
2.4 ✌ 重要参数max_iter & tol
在之前描述K-Means的基本流程时我们提到过,当质心不再移动,Kmeans算法就会停下来。但在完全收敛之前,
我们也可以使用max_iter,最大迭代次数,或者tol,两次迭代间Inertia下降的量,这两个参数来让迭代提前停下
来。有时候,当我们的n_clusters选择不符合数据的自然分布,或者我们为了业务需求,必须要填入与数据的自然
分布不合的n_clusters,提前让迭代停下来反而能够提升模型的表现。
max_iter:整数,默认300,单次运行的k-means算法的最大迭代次数
tol:浮点数,默认1e-4,两次迭代间Inertia下降的量,如果两次迭代之间Inertia下降的值小于tol所设定的值,迭
代就会停下
1.2.2 ✌ DBSCAN算法的基本原理
DBSCAN算法可以看作是一种密度聚类,就是根据样本点分布密度去进行簇的分类
DBSCAN算法核心:
- 随机选取一个未分类的样本点
- 以该样本点为圆心,按照设定的半径画圆,如果该圆中的样本数达到设定的阈值,则将器归为一类,否则舍弃
- 选定该圆中的其他样本点再次进行画圆,直到用光该类中的所有样本点
- 再次选取一个未分类的样本点,重复2、3步骤,直到没有样本点可用
- 如果算法完毕后,仍有点未进行分类,则将其视为离群点
#采用 DBSCAN from sklearn.cluster import DBSCAN dbs=DBSCAN(min_samples=5) dbs.fit(x) label_dbs=dbs.labels_ color=['red','green','yellow','blue'] plt.figure(figsize=(6,6)) for i in range(4): plt.scatter(x[label_dbs==i]['特征1'],data[label_dbs==i]['特征2'],c=color[i]) plt.show()
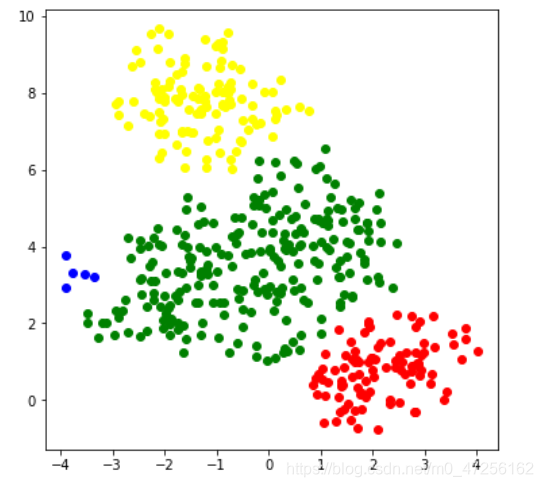
2.1 ✌ DBSCAN重要参数eps,min_samples
eps:该参数为画圆时的半径
min_samples:该参数为圆内最小样本数
1.3 ✌ 簇内平方和的定义
我们分类依据就是认为各个簇内的数据有相似性,而不同簇内的数据是不一样的。我们就会追求“簇内差异小,簇外差异大”,那么这个我们就可以用样本点到其所在中心的距离来衡量。
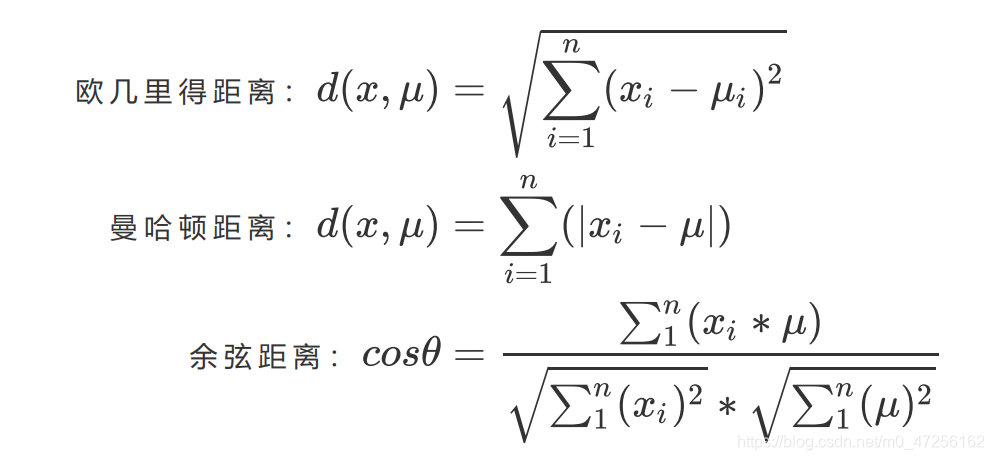
如果我们采用欧几里得距离,则一个簇中样本点到中心距离的平方和为:

第一个公式就是簇内平方和,又叫做Inertia
将所有簇的平方和相加就会的到整体平方和
大家可以发现,Inertia是基于欧几里得距离的计算公式得来的。实际上,我们也可以使用其他距离,每个距离都有自己对应的Inertia。不同距离所对应的质心选择方法和Inertia,在Kmeans中,只要使用了正确的质心和距离组合,无论使用什么样的距离,都可以达到不错的聚类效果
1.4 ✌ 模型评估标准
对于一些分类模型,他们有明确的标签,我们可以用已知的标签和预测结果进行处理获得模型的分数,而聚类没有明确的标签,我们不知道模型的分类如何,而且聚类结果也是不确定的
上面说到聚类是保证“簇内差异小,簇外差异大”,我们可以通过过Inertia来评估簇内差异指标,如果该距离越小,说明模型越好
但是优缺点,这个模型没有界,我们很难找到一个区间来评估出模型的好坏,只能看出相对的好和坏,而且当数据维数过大时,计算量非常大,另外他会随着k的值的变化而变化,比如说我们将k定义为样本数量,那么簇内距离肯定是0,随着k增大,Inertia肯定会减小,但是这并不代表模型的效果会变得更好
所以可以利用一下几个模型评估标准:轮廓系数,calinski_harabasz_score,Homogeneity, completeness and V-measure
1.4.1 ✌ 轮廓系数
在99%的情况下,我们是对没有真实标签的数据进行探索,也就是对不知道真正答案的数据进行聚类。这样的聚
类,是完全依赖于评价簇内的稠密程度(簇内差异小)和簇间的离散程度(簇外差异大)来评估聚类的效果。其中
轮廓系数是最常用的聚类算法的评价指标。它是对每个样本来定义的,它能够同时衡量:
1)样本与其自身所在的簇中的其他样本的相似度a,等于样本与同一簇中所有其他点之间的平均距离
2)样本与其他簇中的样本的相似度b,等于样本与下一个最近的簇中的所有点之间的平均距离
根据聚类的要求”簇内差异小,簇外差异大“,我们希望b永远大于a,并且大得越多越好。
单个样本的轮廓系数计算为:

这个公式可以被解析为:

很容易理解轮廓系数范围是(-1,1),其中值越接近1表示样本与自己所在的簇中的样本很相似,并且与其他簇中的样
本不相似,当样本点与簇外的样本更相似的时候,轮廓系数就为负。当轮廓系数为0时,则代表两个簇中的样本相
似度一致,两个簇本应该是一个簇。可以总结为轮廓系数越接近于1越好,负数则表示聚类效果非常差。
如果一个簇中的大多数样本具有比较高的轮廓系数,则簇会有较高的总轮廓系数,则整个数据集的平均轮廓系数越
高,则聚类是合适的。如果许多样本点具有低轮廓系数甚至负值,则聚类是不合适的,聚类的超参数K可能设定得
太大或者太小。
在sklearn中,我们使用模块metrics中的类silhouette_score来计算轮廓系数,它返回的是一个数据集中,所有样
本的轮廓系数的均值。但我们还有同在metrics模块中的silhouette_sample,它的参数与轮廓系数一致,但返回的
是数据集中每个样本自己的轮廓系数。
from sklearn.metrics import silhouette_score score_sil=silhouette_score(x,label_kms)
1.4.2 ✌ Calinski_harabasz_score
卡林斯基-哈拉巴斯指数(Calinski-Harabaz Index,简称CHI,也被称为方差
比标准),戴维斯-布尔丁指数(Davies-Bouldin)以及权变矩阵(Contingency Matrix)可以使用。

其中N为数据集中的样本量,k为簇的个数(即类别的个数), 是组间离散矩阵,即不同簇之间的协方差矩阵,
是簇内离散矩阵,即一个簇内数据的协方差矩阵,而tr表示矩阵的迹。在线性代数中,一个n×n矩阵A的主对角
线(从左上方至右下方的对角线)上各个元素的总和被称为矩阵A的迹(或迹数),一般记作 。数据之间的离
散程度越高,协方差矩阵的迹就会越大。组内离散程度低,协方差的迹就会越小, 也就越小,同时,组间
离散程度大,协方差的的迹也会越大, 就越大,这正是我们希望的,因此Calinski-harabaz指数越高越好。
该评估标准总之为越大越好
from sklearn.metrics import calinski_harabasz_score score_cal=calinski_harabasz_score(x,label_kms)
该评估标准相对轮廓系数来说有个巨大的有点就是计算速度快
1.4.3 ✌ Homogeneity, completeness and V-measure
from sklearn.metrics import homogeneity_score from sklearn.metrics import completeness_score from sklearn.metrics import v_measure_score hom_score=homogeneity_score(y,label_kms) com_score=completeness_score(y,label_kms) v_score=v_measure_score(y,label_kms) v_score
