
基因分析平台中DNAseq.SentieonFastqToVcf成功运行但结果不对
为什么按默认参数运行后只分析了上传fastq中的2万个reads
?
并且运行15分钟就任务完成了。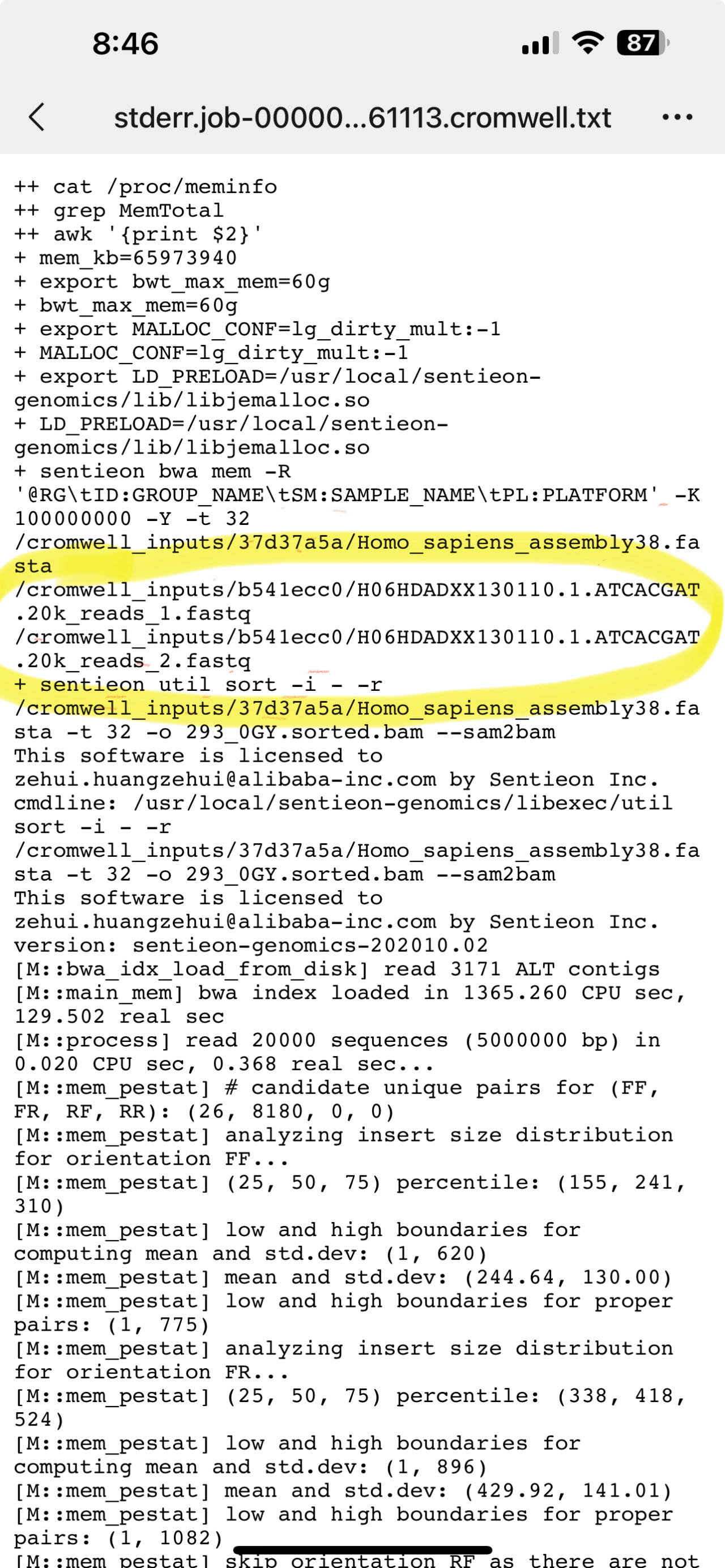
展开
收起
版权声明:本文内容由阿里云实名注册用户自发贡献,版权归原作者所有,阿里云开发者社区不拥有其著作权,亦不承担相应法律责任。具体规则请查看《阿里云开发者社区用户服务协议》和《阿里云开发者社区知识产权保护指引》。如果您发现本社区中有涉嫌抄袭的内容,填写侵权投诉表单进行举报,一经查实,本社区将立刻删除涉嫌侵权内容。
1
条回答
写回答
-
面对过去,不要迷离;面对未来,不必彷徨;活在今天,你只要把自己完全展示给别人看。
在您使用DNAseq.SentieonFastqToVcf运行基因分析平台时,遇到的首个问题是虽然程序成功运行,但是结果却不准确。这个问题可能的原因是您上传的FASTQ文件中只包含了2万个reads,这导致在将reads映射到参考基因组的过程中,可能没有足够的数据进行分析。为了解决这个问题,您可以考虑增加上传的reads数量,或者调整程序中的参数设置,比如使用-L参数来指定特定的染色体或者基因组区域进行测序和分析。
关于运行时间过短的问题,有可能是因为默认参数设置较为优化,所以在短时间内就完成了任务。然而,如果您觉得15分钟的任务完成时间过短,可能需要调整一些参数以增加程序的运行时间。例如,Sentieon软件提供了vcfconvert模块来压缩所有的gvcf文件并建立索引,通过这种方式可以显著提高处理速度。此外,您也可以考虑加大程序运行时的线程数(如-t参数所设定)来进一步延长处理时间。
2024-01-16 15:13:58赞同 展开评论

相关问答
热门讨论
热门文章
展开全部
还有其他疑问?
咨询AI助理