引言
本系列开启R中单细胞RNA-seq数据分析教程,持续更新,欢迎关注,转发!
简介
现在,很少有人只进行一次单细胞RNA测序实验并仅产生一份数据。原因很直接:目前的单细胞RNA测序技术每次只能捕捉到有限样本的分子状态。为了在多个实验和不同条件下对众多样本进行测量,通常需要对来自不同实验的单细胞RNA测序数据进行联合分析。虽然有些实验策略,比如 细胞哈希!,以及一些计算方法,比如 demuxlet 和 scSplit,能够在一定程度上将多个样本合并进行单细胞RNA测序库的构建和测序,但像组织分离这样的步骤仍然需要针对不同样本单独进行。因此,与处理常规RNA-seq数据一样,批次效应往往是需要解决的关键干扰因素。
在本教程将介绍几种单细胞RNA测序数据整合的方法。需要记住的是,目前还没有一种整合方法能够适用于所有情况。因此,尝试多种方法并进行比较,最终选择最适合特定情况的方法,是非常重要的。
0. 数据加载
从导入Seurat并加载保存的Seurat对象开始。
library(Seurat)
library(dplyr)
library(patchwork)
seurat_DS1 <- readRDS("DS1/seurat_obj_all.rds")
seurat_DS2 <- readRDS("DS2/seurat_obj_all.rds")
1. 合并这两个数据集
首先,有可能批次效应并不显著,因此不需要进行特别的整合。所以,应当首先尝试将这两个数据集直接合并,以便进行初步的观察。
seurat <- merge(seurat_DS1, seurat_DS2) %>%
FindVariableFeatures(nfeatures = 3000) %>%
ScaleData() %>%
RunPCA(npcs = 50) %>%
RunUMAP(dims = 1:20)
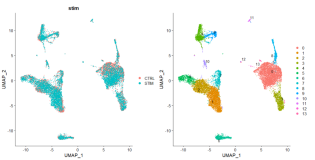
plot1 <- DimPlot(seurat, group.by="orig.ident")
plot2 <- FeaturePlot(seurat, c("FOXG1","EMX1","DLX2","LHX9"), ncol=2, pt.size = 0.1)
plot1 + plot2 + plot_layout(widths = c(1.5, 2))

显然,这两个数据集在嵌入图上是相互独立的。然而,标记基因的表达模式显示,这两个数据集实际上有许多共同的细胞类型。理想情况下,两个数据集中相同类型的细胞应该能够混合在一起。但由于批次效应的影响,它们并没有混合。因此,需要对数据进行整合。的目标是,在整合之后,两个数据集中相同类型的细胞能够混合,而不同类型的细胞或状态仍然保持分离。
将尝试以下几种不同的整合方法:
- Seurat
- Harmony
- LIGER
- MNN
- RSS to BrainSpan
- CSS
2.1. 使用Seurat进行数据整合
Seurat内置了数据整合流程。简单来说,它首先对需要整合的数据集执行典型相关分析(CCA),独立旋转它们以最大化两数据集之间的协方差。换言之,Seurat利用CCA寻找增强数据集间相似性的最佳方式。之后,Seurat引入了一个锚定机制,在两个数据集中寻找细胞锚点。细胞锚点由来自不同数据集的一对细胞组成,它们在CCA空间中互为最近邻,同时一个细胞在自己数据集中的最近邻也倾向于与另一细胞的最近邻相邻。这两个锚定的细胞被视为两个数据集中相互对应的细胞,然后通过计算两个数据集中锚定细胞对的转换矩阵,并从其中一个数据集的表达中减去这个转换矩阵来执行整合过程。
使用Seurat进行整合前,需要先对每个待整合的数据集进行归一化并识别高变异基因(这应该是已经完成的步骤)。如果尚未完成,需要先进行这一步:
seurat_DS1 <- NormalizeData(seurat_DS1) %>% FindVariableFeatures(nfeatures = 3000)
seurat_DS2 <- NormalizeData(seurat_DS2) %>% FindVariableFeatures(nfeatures = 3000)
然后,确定数据集的锚点。在此步骤中,Seurat 需要一个包含多个 Seurat 对象的列表作为输入。需要指出的是,Seurat 支持整合两个以上样本的数据。只需将它们列成一个列表即可。
seurat_objs <- list(DS1 = seurat_DS1, DS2 = seurat_DS2)
anchors <- FindIntegrationAnchors(object.list = seurat_objs, dims = 1:30)
然后,把确定的锚点集合输入到 IntegrateData 函数中,以便对表达水平进行校正。
seurat <- IntegrateData(anchors, dims = 1:30)
执行 IntegrateData 函数会生成一个新的 Assay 对象(默认名称为 integrated),其中保存了经过批次校正后的表达矩阵。原始未经校正的数据仍然保留,在默认名为 RNA 的原始 Assay 对象中。生成的 Seurat 对象默认的测定方法会被设置为 integrated,但可以通过代码 DefaultAssay(seurat) <- "RNA" 切换回默认的测定方法。
然后,只需使用校正后的 Seurat 对象,并重新执行第一部分的流程,但需要跳过前两个步骤,即归一化和识别高变异基因这两个步骤。
seurat <- ScaleData(seurat)
seurat <- RunPCA(seurat, npcs = 50)
seurat <- RunUMAP(seurat, dims = 1:20)
seurat <- FindNeighbors(seurat, dims = 1:20) %>% FindClusters(resolution = 0.6)
# You may also want to save the object
saveRDS(seurat, file="integrated_seurat.rds")
需要提醒的是,尽管 tSNE/UMAP 的嵌入和聚类分析应当基于 integrated 测定来进行,但校正后的数据作为基因表达量的定量指标已不再十分可信。因此,建议在进行其他分析,例如识别聚类标记和进行可视化时,改用未经校正的表达值,方法是将 DefaultAssay 切换回 RNA。
DefaultAssay(seurat) <- "RNA"
plot1 <- UMAPPlot(seurat, group.by="orig.ident")
plot2 <- UMAPPlot(seurat, label = T)
plot3 <- FeaturePlot(seurat, c("FOXG1","EMX1","DLX2","LHX9"), ncol=2, pt.size = 0.1)
((plot1 / plot2) | plot3) + plot_layout(width = c(1,2))

虽然它不是完美的,但它确实有助于提高两个数据集的可比性。
如果你想进一步提升结果,有几个参数你可以考虑调整(所有上述参数要么是默认设置,要么是基于直觉,所以应该还有改进的空间)。首先,FindIntegrationAnchors 函数根据它们在各个数据集中被识别为高变异基因的频率来选择用于整合的基因。因此,在两个数据集上执行 FindVariableFeatures 时的 nfeatures 参数肯定会影响用于整合的基因集。接下来,由于锚定步骤是 Seurat 整合中的关键步骤,任何显著影响锚定过程的参数都可能改变最终的整合结果。例如,FindIntegrationAnchors 函数默认选择2000个在各个数据集中被高频识别为高变异基因的基因用于整合,这个用于整合的基因数量可以通过在 FindIntegrationAnchors 函数中设置 anchor.features 参数来调整。类似于决定使用多少 PCs 来制作 tSNE/UMAP 和聚类的问题,需要决定使用哪些 CCs 来定义跨数据集的邻居,如在 dims 参数中设置。这是另一个可以影响结果的参数。在同一函数中还有更多参数可以影响结果,包括 k.anchor、k.filter 和 k.score,尽管它们可能不是你想首先调整的参数。类似地,在下一步使用的 IntegrateData 函数中也有 dims 参数,你可能也想改变它。
值得一提的是,Seurat 还提供了另一种整合分析策略,即数据转移。当存在一个已注释的参考数据集,并且想要使用参考数据来辅助新查询数据的细胞类型/状态注释时,就会使用到它。数据整合和数据转移之间的主要区别包括:
- 与数据整合时使用 CCA 生成一个联合空间不同,数据转移默认将参考数据中的相同 PCA 转换应用于查询数据集以识别锚点。
- 不校正任何表达值,因此不会创建两个数据集的联合嵌入;相反,可以将查询数据中的细胞投影到参考嵌入中。除了嵌入,还可以投影细胞标签,以便可以将参考图谱中的 '转移' 标签到查询数据集中进行注释。
2.2. 使用 Harmony 进行数据整合
除了 Seurat,现在还有更多数据整合方法可用。由 Soumya Raychaudhuri 实验室开发的 Harmony 就是其中之一。它也是第一个关于 scRNA-seq 批次效应校正工具的 基准测试中最受关注的整合方法。简而言之,Harmony 使用模糊聚类将每个细胞分配给多个聚类。对于每个聚类,它然后计算每个数据集的校正因子,将该数据集的聚类中心向该聚类的全局中心移动。由于每个细胞被表示为多个聚类的组合,因此通过平均细胞所属聚类的校正因子,并按聚类分配比例加权,计算出细胞特定的校正因子。这个过程将迭代进行,直到收敛发生或达到迭代限制。
Harmony 为 Seurat 对象提供了一个简单的 API,即一个名为 RunHarmony 的函数,因此非常容易使用。它接受合并后的 Seurat 对象(在第1步生成的那个)作为输入,并且需要告诉函数使用哪个元数据特征作为批次身份。它返回一个 Seurat 对象,并添加了一个名为 harmony 的更多校正。它就像校正后的 PCA,因此应该明确告诉 Seurat 在后续分析中使用 harmony 校正,包括制作 UMAP 嵌入和识别细胞聚类。
seurat <- merge(seurat_DS1, seurat_DS2) %>%
FindVariableFeatures(nfeatures = 3000) %>%
ScaleData() %>%
RunPCA(npcs = 50)
library(harmony)
seurat <- RunHarmony(seurat, group.by.vars = "orig.ident", dims.use = 1:20, max.iter.harmony = 50)
seurat <- RunUMAP(seurat, reduction = "harmony", dims = 1:20)
seurat <- FindNeighbors(seurat, reduction = "harmony", dims = 1:20) %>% FindClusters(resolution = 0.6)
# You may also want to save the object
saveRDS(seurat, file="integrated_harmony.rds")
接下来,可以按照之前的方法来展示整合后的结果。
plot1 <- UMAPPlot(seurat, group.by="orig.ident")
plot2 <- UMAPPlot(seurat, label = T)
plot3 <- FeaturePlot(seurat, c("FOXG1","EMX1","DLX2","LHX9"), ncol=2, pt.size = 0.1)
((plot1 / plot2) | plot3) + plot_layout(width = c(1,2))

结果还算不错。两个样本的细胞混合得很好,可以观察到一些清晰的轨迹。对于一些混合的细胞群体,特别是非背侧端脑的细胞,需要进一步确认它们是否真的是应该混合的相同类型的细胞。
你可能已经注意到,Harmony 默认使用 PCA 结果作为输入,并针对每个细胞的 PCs 进行校正迭代。因此,影响原始 PCA 的参数,比如在 FindVariableFeatures 中用于识别高变异基因的 nfeatures,可能会对整合结果产生影响。如果没有提供特定的参数,RunHarmony 函数将默认使用输入中的所有可用维度(通常是 PCA)。可以通过设置 dims.use 参数来指定使用哪些维度(这个参数与 Seurat 中的许多其他 dims 参数类似)。