凯凯何_Boy

已加入开发者社区1790天
勋章

专家博主
专家博主

星级博主
星级博主

技术博主
技术博主

初入江湖
初入江湖



我关注的人
粉丝

技术能力
兴趣领域
擅长领域
技术认证
暂时未有相关云产品技术能力~
暂无个人介绍
暂无精选文章
暂无更多信息
2023年06月
-
06.16 08:20:47
 发表了文章
2023-06-16 08:20:47
发表了文章
2023-06-16 08:20:47
R语言绘制组间比较散点图并自动添加P值信息
查询ggprism包使用时候发现官网给出的一示例图比较常用,这里记录学习一下。 -
06.16 08:17:12发表了文章
2023-06-16 08:17:12
利用abbrevr包批量输出期刊缩写
有时候用endnote导入文献后显示的是期刊全称,而用到缩写时候就需要去一些网站上一个个搜索,比如CASSI, LetPub、Pubumed等网站,或者Y叔公号里直接回复,而逛Github时候突然发现abbrevr这小R包中可以很快批量实现这个需求,在此记录一下。 -
06.16 08:14:11发表了文章
2023-06-16 08:14:11
利用massdatabase包提取物种KEGG通路与基因/化合物对应信息
最近手头处理一批代谢组数据, 想基于几十个关键差异代谢物代谢物进行下KEGG富集,能想到有两种方式解决,一种常用方式就是基于MetaboAnalyst在线富集,另一种就是解析出该物种的通路与代谢物的对应关系文件,然后用Y叔叔的Clusterprofiler包富集。经一番搜索,massdatabase包可帮我们轻松获得这个文件。 作者:凯凯何_Boy 链接:https://www.jianshu.com/p/654784925903 来源:简书 著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。 -
06.16 08:08:12发表了文章
2023-06-16 08:08:12
基于GO-Figure快速绘制GO富集气泡图
GO-Figure! 是一个基于pyhton写的GO富集小软件,方便我们拿到富集结果后直接使用命令行的形式进行可视化,绘制出具有灵活性、可重复性的图形,且可基于最新的GO数据库内容进行富集。效果图如下,若用惯了常见的气泡柱形图,这种形式也不失为一种新颖的可视化方法~ -
06.15 22:54:28发表了文章
2023-06-15 22:54:28
一个轻量级绘制热图的R包-HeatmapR
目前R中绘制热图的方式有很多,常用的如pheatmap、ComplexHeatmap包等,这里再给大家介绍一个轻量级的R包-HeatmapR包,即无需过多的前期数据处理,可同时可视化含有离散型和连续性的矩阵。 -
06.15 22:51:23发表了文章
2023-06-15 22:51:23
分级KEGG富集pathway图绘制
最近在基迪奥平台上看到了这张KEGG富集信息图,刚好手头有批现成的转录组数据,刚好绘制一下给富集图换换风格~~ -
06.15 22:46:56发表了文章
2023-06-15 22:46:56
学会这三个包,搞定R中的正则
天下苦正则久矣,尤其当字符串中有自己想要的一段数据时候苦苦提取不出来的感觉真是难受,今天给大家介绍3个R包来解决这种正则带来烦恼的包,希望对大家能有所帮助。 -
06.15 22:41:56发表了文章
2023-06-15 22:41:56
SFINX: 一个基于Shiny部署的鉴定蛋白互作关系平台
目前研究蛋白质互作方法有很多,传统的方法是将天然蛋白免疫沉淀与质谱检测结合(CoIP-MS),另外流行的还有亲和纯化/质谱法(AP-MS),与CO-IP类似,它使用感兴趣的诱饵蛋白(bait proteins)上的表位标签和捕获探针来识别协同的猎物蛋白,不需要为每个新的诱饵蛋白购买或者开发特定抗体,得到的融合蛋白可以用链霉亲和素(strep)磁珠来亲和纯化,用生物素洗脱最终得到蛋白复合物。 -
06.15 22:37:27发表了文章
2023-06-15 22:37:27
微生物组(16S rRNA)数据分析套件PMS尝鲜
前几日宏基因组公号上推送了《iMeta:青岛大学苏晓泉组开发跨平台可交互的微生物组分析套件PMS》一文,但软件中示例文件貌似是单端数据,于是想着拿手里一批已发表过的双端测序16S数据集尝试一番。 -
06.15 22:30:51发表了文章
2023-06-15 22:30:51
Robust火山图:一种含离群值的代谢组数据差异分析方法
代谢组学中差异代谢物的识别仍然是一个巨大的挑战,并在代谢组学数据分析中发挥着突出的作用。由于分析、实验和生物的模糊性,代谢组学数据集经常包含异常值,但目前可用的差异代谢物识别技术对异常值很敏感。作者这里提出了一种基于权重的具有稳健性火山图方法,助于从含有离群值的代谢组数据中更加准确鉴定差异代谢物。 -
06.15 22:26:51发表了文章
2023-06-15 22:26:51
GSEA会用了,来了解下DGSEA?
差异基因集富集分析(DGSEA)是对Broad Institute / MIT的原始的Gene Set Enrichment Aanlysis(GSEA)分析算法的改进。它是一种用来确定输入基因集在两种生物通路之间是否显示出统计学上差异计算方法。 -
06.15 22:23:01发表了文章
2023-06-15 22:23:01
MonaGO:一款新颖的交互式富集分析web工具
Monash Gene Ontology (MonaGO) 是一种新颖的基于 Web 的可视化系统,它提供了一个直观、交互式和响应式的界面,用于执行 GO 富集分析和可视化结果。MonaGO支持基于DAVID富集结果可视化,或者直接输入GO terms进行可视化。 -
06.15 22:14:28发表了文章
2023-06-15 22:14:28
基于R语言绘制Network几种方式
平时对于网络图的绘制,一般我们都会在R中生成边和点列表后导入到Cytoscape和Gephi等的本地工具软件当中,而R语言中也自带有不少优秀的包也可精美的可视化我们的数据,所有函数也比较简单,有时间的不妨学习一下~~ -
06.15 21:23:09发表了文章
2023-06-15 21:23:09
利用MAGeCK算法处理CRISPR Screen数据
上次文章结尾时候提到了MAGeCK RRA算法处理,这次我们就来学习一下,Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout(MAGeCK) 是一个可以从全基因组CRISPR-CAS9筛查技术中识别重要基因计算工具。Mageck是由Wei Li 和 Shirley Liu lab共同开发维护的。 -
06.15 21:16:02发表了文章
2023-06-15 21:16:02
一条命令行区分Contigs中的真核原核序列
本文介绍一款可用于宏基因组中的分类小软件,简单一条命令可以将上游组装的Contigs进行原核与真核生物区分~ -
06.15 21:13:26发表了文章
2023-06-15 21:13:26
DiVenn一款用于比较基因列表的交互式Venn图绘制工具
Divenn一种基于网络的基于Web的工具 Divenn 可以将来自多个RNA-seq实验中的基因列表进行比较,并显示每个基因(集)的调控水平和整合到Kegg pathway和GO term的数据信息。我们通过该工具更加方便的了解重叠基因与其相关的pathway或Go terms之间的表达模式,十分有趣。 -
06.15 21:03:15发表了文章
2023-06-15 21:03:15
基于R筛选过滤低丰度物种的几种方式
基于R筛选过滤低丰度物种的几种方式 -
06.15 20:58:16发表了文章
2023-06-15 20:58:16
ggpattern包-基于几何图案或图像的自定义填充
平时我们做柱状图或饼图都会用彩色进行填充,但是文章有时候为了节约成本采用黑白印刷时候,图形一般都会做成各种阴影线条填充模式来进行区分(如下图),R中的ggpattern包刚好可以满足了我们的需求,若有需要就来学习下吧~ -
06.15 20:47:53发表了文章
2023-06-15 20:47:53
R可视化学习-- (8) 环形柱状图
柱状图可谓是论文最常见的图表了,但有时候这种笔直排列一排的柱状图用腻了, 换种呈现方式(比如拐个弯)也可换个心情嘛,今天就先来介绍俩种构建环形柱状图的方法,分别基于ggplot2和其他的R包来绘制。 -
06.15 20:44:17发表了文章
2023-06-15 20:44:17
如何使折线图的线条纵享丝滑?
昨天在做一个简单的频率分布直方图时,想在上方增添一个折线图,但是发现简单的geom_line()所添加的曲线有点生硬,在想有没有使其平滑的方法,于是google一番发现还是很容易实现的~~ -
06.15 20:41:10发表了文章
2023-06-15 20:41:10
利用TCseq包进行基因表达趋势分析
TCseq包提供了一个统一的套件去处理不同时序类型的数据分析,可以应用于转录组或者像ATAC-seq,Chip-seq的表观基因组时序型数据分析。该包主要的集中于不同时间点的差异分析,时间趋势分析及可视化作图。 -
06.15 20:30:21发表了文章
2023-06-15 20:30:21
让你的Typora笔记自动生成思维导图
现在应该许多人喜欢用Markdown方式去做笔记,其中最流行的软件当属Typora了,因为功能比较齐全,且Typora中提供了大纲的功能可以在笔记左侧清晰展示内容中各个大标题,方便我们查看。 -
06.15 20:24:48发表了文章
2023-06-15 20:24:48
利用ggcor包绘制相关性组合图及环状热图
ggcor包最初是因为能快速实现19年Science一组合相关性图(上图所示)变得流行起来,除此该包对热图、热图等等的可视化都是很方便快捷的,除了之前介绍过的几种相关性图几种方式,此包也是个不错的选择,且具独特的风格(特别是组合相关性图、环形热图)。但是不知道因为何种原因此包在Github上消失了....,到作者(厚缊)个人博客上瞅了瞅发现关于包的参数介绍示例等也都没有了,在评论区里看到作者回答项目已不再提供任何代码和任何资料,需要的可以去国内的gitee和国外的github搜索看看有没有别人存的代码。 -
06.15 20:16:11发表了文章
2023-06-15 20:16:11
R可视化学习(7) -- 气泡图
气泡图 -
06.15 20:00:13发表了文章
2023-06-15 20:00:13
Fastqc用腻了,来试下这个R包吧
平时拿到数据后首先要进行质控环节,其中FastaQC软件的使用最为广泛,它可以为每一个样品生成一个html报告和一个‘zip’ 文件,zip解压之后生成fastqc_data.txt和summary.txt的文件,里面包含了测序样品的质量信息,但是如果有几十个甚至上百个数据时候,我们总不可能一一的打开每个html文件查看,因此Multiqc软件成为了解决这一问题的首选方案,可以基于结果zip文件进行多样品的整合分析,信息生成在一张报告上。然而哪里有问题,哪里就有R包,对于R语言爱好者,这里再提供一种可替代的方法--fastqcr包,好处就是不管有没有linux环境时候,都可以快速对数据进行质检 -
06.15 19:51:20发表了文章
2023-06-15 19:51:20
R可视化学习(5) -- 脊线图
Ridgeline 图(脊线图),(有时称为Joyplot)可以同时显示几个组的数值分布情况,分布可以使用直方图或密度图来表示,它们都与相同的水平尺度对齐,并略有重叠。常常被用来可视化随时间或空间变化的多个分布/直方图变化。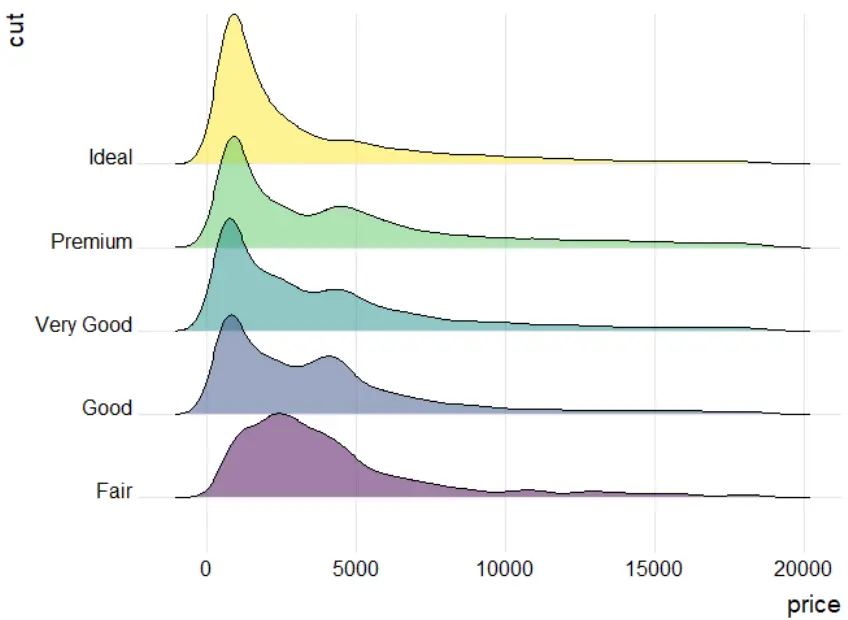
-
06.15 19:45:50发表了文章
2023-06-15 19:45:50
R可视化学习(4) -- 棒棒糖图
棒棒糖图其实类似于柱状图加散点图的效果,因为他的形状就是由俩部分组成(点+线条),因此在ggplot中,我们只要通过geom_point()函数绘制"糖"的那一部分,geom_segment()函数绘制“棒棒”那一部分,就可轻松绘制出这种图形 -
06.15 19:39:11发表了文章
2023-06-15 19:39:11
Brename - 一个便捷跨平台批重命名文件/目录的命令行小工具
brename是用Go编程语言实现的,支持跨平台Linxu和Windows用户使用。 -
06.15 19:30:21发表了文章
2023-06-15 19:30:21
宏基因组之基因组装
宏基因组组装,即把短的reads拼装成连续的序列contig,再根据PE等关系将contig拼装成scaffold。与单个基因组组装不同,宏基因组组装最终得到的是环境样品中全部微生物的混合scaffold。理想情况下一条scaffold对应一个物种的全基因组。但由于序列太短或者覆盖度不够,很难拼出一条完整的基因组。针对高通量测序数据,出现了多种拼接算法和软件。 -
06.15 19:23:55发表了文章
2023-06-15 19:23:55
R可视化学习(3)--误差棒图
篇介绍如何利用R软件和ggplot2制作误差线的条形图,我们可以使用如下几个函数制作不同类型的误差线图形: • geom_errorbar() • geme_linerange() • geom_pointrange() • geom_crossbar() • geom_errorbarh() -
06.15 18:51:02发表了文章
2023-06-15 18:51:02
盘一盘画相关性热图的几种方式
R可视化相关性矩阵的几种方案 R中相关性矩阵的可视化解决方法在已经有很多了,我们在这里总结一些常用的都有哪些: 1. ggplot2包 2. corrplot包 3. 热图包(pheatmap,heatmap包等) 4. ggally 包中的 ggcorr() 函数 5. ggcorrplot 包 -
06.15 18:37:46发表了文章
2023-06-15 18:37:46
CORNAS:一种快速简单鉴定无重复转录组差异基因的方法
还记得上次文章的最后提到CORNAS这种方法吗?最近刚好在Github上看到了这个项目,就花了点时间看了下文档感觉操作也比较简单,这里记录一下使用过程,大家共同学习一下。 -
06.15 18:30:04发表了文章
2023-06-15 18:30:04
没有生物学重复的转录组数据怎么进行差异分析?
设置生物学重复这个环节也是你实验设计很重要的一part,设置的好对你下游分析也有利,通常我们做转录组测序,需要的样本量每组至少为3个生物学重复,这个处理起来就很合理,并且现在流行的差异分析软件DEseq2,limma,edgeR等等都是针对有重复的数据去做的,但有时候会不幸碰到样品测序失败不能用,导致每组就给你剩一个重复时候该怎么办,之前我有批数据就是这样,但是办法总比困难多不能放过任何实验数据,搜了搜其实还是有一些方法可以去解决的,在这里介绍下我搜到的几种方法。 -
06.15 18:23:00发表了文章
2023-06-15 18:23:00
gg.gap包—截断Y轴小能手
截断Y轴往往是我们作柱形图时候,当有一组数据的分布过大或者过小时候需要用到,不截断的话值小的变量信息往往会被掩盖,图片也会不美观,今天介绍俩个方便截断Y轴方法供大家参考。 -
06.15 18:12:43发表了文章
2023-06-15 18:12:43
谈谈转录组测序基础知识及常见问题
转录组学(Transcriptomics),是一门在真整体水平上研究细胞中基因转录的情况及转录调控规律的学科,从RNA水平研究基因的表达情况。转录组测序是通过二代测序平台快速全面地获得某一物种特定细胞或组织在某一状态下的几乎所有的转录本及基因序列,可以用来研究基因表达量、基因功能、结构、可变剪接和预测新的转录本等等。转录组(transcriptome),是指特定生长阶段某组织或细胞内所有转录产物的集合,狭义上指所有mRNA的集合。 -
06.15 17:52:26发表了文章
2023-06-15 17:52:26
R可视化学习—词云图
词云。又称文字云。“词云”就是通过形成“关键词云层”或“关键词渲染”,对网络文本中出现频率较高的“关键词”的视觉上的突出,它会过滤掉大量的文本信息,使浏览网页者只要一眼扫过文本就可以领略文本的主旨。 -
06.15 17:43:05发表了文章
2023-06-15 17:43:05
Bingo(Cytoscape插件)GO功能富集分析
转录组分析中基因的GO富集分析这步有很多软件可以选择,这次我们来介绍下如何利用Cytoscape软件中的一个简单又好用的Bingo小插件完成富集的工作,Cytoscape其实是一个做网络图的软件,所以使用Bingo的这种方法特别之处就在于富集的结果是可以直接以网络图的形式呈现,类似于下图这种效果,区别于其他的软件大多只生成表的形式。 -
06.15 17:33:06发表了文章
2023-06-15 17:33:06
WEGO使用—在线GO功能注释
WEGO是华大基因公司的一个在线GO注释的网站,根据网站的介绍:WEGO(网络基因本体注释图)是一个简单但有用的工具,用于可视化,比较和绘制基因本体注释结果。随着围棋词汇越来越流行,WEGO在许多研究中被广泛采用和使用,最新一次的更新是在2018年。 -
06.15 16:56:47发表了文章
2023-06-15 16:56:47
基于excel和python拆分数据为多行
在基因富集操作后我们会得到这样的结果,像下图这样可以看到富集的通路中都涉及了哪些基因ID,但是这样查看并不友好,我们可以拿这两列数据去做个转换,清楚直观的呈现这种对应关系。也就是将后面按 / 分割的基因拆分为多行。 -
06.15 16:49:35发表了文章
2023-06-15 16:49:35
vscode中粘贴文本缩进混乱的解决
有时候用vscode写代码时候,会碰到这样的问题,从Excel表中复制制表符分割的文件到vscode中去进行处理,分隔符就变了,像下图一样,有的行1个空格分割,有的行变为3个空格,格式已经发生混乱。这样会导致写代码用split分割时候你不知道以什么分割,文本少的时候还OK,手动改改,文本大的时候,就麻烦了必须得让他们统一,复制的啥格式粘贴就得是啥格式。 -
06.15 16:47:15发表了文章
2023-06-15 16:47:15
Windows下如何使用tree命令生成目录树
熟悉Linux的人应该对tree命令不陌生,可以使我们对指定目录制作一种目录树的形式,就像下面这种形式。 -
06.15 16:42:53发表了文章
2023-06-15 16:42:53
为你的项目做一份Rmarkdown报告吧
markdown是一种轻量级标记语言,现在许多软件例如Mou、MarkdownEditor、Haroopad、Typora等,通过这些工具可以便捷的完成markdown文字录入,并且支持导出PDF、HTML等格式。对markdown语法还不太了解的人,请自行百度了解,个人认为只要花上几个小时你就能掌握,确实没什么难度,本文主要简单介绍R环境中的markdown,也就是Rmarkdown这个包怎么一步步的制作我们的项目报告。 -
06.15 16:35:17发表了文章
2023-06-15 16:35:17
利用ggpubr绘制双坐标轴
之前画双Y轴的图都是自己写函数用ggplot2去制作,偶然间发现了ggpubr包一种可视化的形式,大家可以一起学习下~ -
06.15 16:33:01发表了文章
2023-06-15 16:33:01
利用gghighlight包高亮你的图形
如何在利用ggplot高亮出你想要展示的数据呢?gghighlight包,快速解决你的问题~~ -
06.15 14:38:53发表了文章
2023-06-15 14:38:53
R可视化学习(1)--直方图
本篇介绍如何使用R软件和ggplot2包来制作直方图,我们需要用到geom_histgramh函数,也可以用geom_vline函数去增加线条展示平均值。 -
06.15 14:29:38发表了文章
2023-06-15 14:29:38
宏基因组之基因丰度计算
目前有两种方式可计算宏基因组基因的丰度,一种是基于比对,比如bwa,bowtie,soapaligner等主流的比对软件,另一种是不比对快速估计基因丰度,可以用近俩年来流行的salmon软件,这个软件在转录组的数据比对中也经常用到,可以直接计算出原始的Counts值和标准化的TPM值,此外由于是基于非比对,计算的速度得到很大的提升,同时也节省了部分的内存(少了庞大的sam/bam文件),可以说是多快好省,但是目前的高分文章中也还是不少用基于比对的方法去计算宏基因组的基因丰度的,下面我就分别简单介绍一下基于比对的soapaligner和不比对快速估计的samlon俩个软件的操作流程!! -
06.15 14:23:54发表了文章
2023-06-15 14:23:54
R可视化学习(2)--箱线图
箱线图由箱和“须”(whisker)两部分组成。箱的范围是从数据的下四分位数到上四分位数,也就是常说的四分位距(IQR)。箱的中间有一条表示中位数,或者说50%分位数的线。须则是从箱子的边缘出发延伸至1.5倍四分位距内的最远的点。如果图中有超过须的数据点,则其被视为异常值,并以点来表示。如下图使用偏态的数据展示了直方图、密度曲线和箱线图之间的关系。 -
06.15 14:12:31发表了文章
2023-06-15 14:12:31
拿到基因表达矩阵之后的那点事(四)
得到了差异基因并进行了一顿操作可视化后,我们可以开始富集分析了,Don't say so much,要富集当然首推Y叔的成功之作-- Clusterprofiler, 因为我的数据物种比对的牛的基因组,也是属于模式物种,用该包去做富集是更为方便~~ ,当然这个包也不仅仅限于模式物种,开发者当然会考虑的比较全面,提供了几个函数去做非模式物种或无参的富集,后面我也会说到~~ OK,开始富集吧!!
-
发表了文章
2023-06-16
R语言绘制组间比较散点图并自动添加P值信息
-
发表了文章
2023-06-16
利用abbrevr包批量输出期刊缩写
-
发表了文章
2023-06-16
利用massdatabase包提取物种KEGG通路与基因/化合物对应信息
-
发表了文章
2023-06-16
基于GO-Figure快速绘制GO富集气泡图
-
发表了文章
2023-06-15
一个轻量级绘制热图的R包-HeatmapR
-
发表了文章
2023-06-15
分级KEGG富集pathway图绘制
-
发表了文章
2023-06-15
学会这三个包,搞定R中的正则
-
发表了文章
2023-06-15
SFINX: 一个基于Shiny部署的鉴定蛋白互作关系平台
-
发表了文章
2023-06-15
微生物组(16S rRNA)数据分析套件PMS尝鲜
-
发表了文章
2023-06-15
Robust火山图:一种含离群值的代谢组数据差异分析方法
-
发表了文章
2023-06-15
GSEA会用了,来了解下DGSEA?
-
发表了文章
2023-06-15
MonaGO:一款新颖的交互式富集分析web工具
-
发表了文章
2023-06-15
基于R语言绘制Network几种方式
-
发表了文章
2023-06-15
利用MAGeCK算法处理CRISPR Screen数据
-
发表了文章
2023-06-15
一条命令行区分Contigs中的真核原核序列
-
发表了文章
2023-06-15
DiVenn一款用于比较基因列表的交互式Venn图绘制工具
-
发表了文章
2023-06-15
基于R筛选过滤低丰度物种的几种方式
-
发表了文章
2023-06-15
ggpattern包-基于几何图案或图像的自定义填充
-
发表了文章
2023-06-15
R可视化学习-- (8) 环形柱状图
-
发表了文章
2023-06-15
如何使折线图的线条纵享丝滑?
滑动查看更多
暂无更多信息
暂无更多信息