引言
本系列讲解 使用Scanpy分析单细胞(scRNA-seq)数据教程,持续更新,欢迎关注,转发!
简介
本文介绍了一种基于 PCA 的简单数据整合方法,我们将其命名为 ingest(),并且将它与 BBKNN进行了对比。BBKNN 能够很好地融入 Scanpy 工作流程中,可以通过 bbknn() 函数来使用。
ingest() 函数需要一个标注过的参考数据集,这个数据集要能够体现我们感兴趣的生物学变化。它的思路是在参考数据上构建一个模型,然后利用这个模型来处理新的数据。目前,这个模型是由 PCA 加上一个邻居查找搜索树组成的,我们用的是 UMAP 的相关实现。
ingest()方法简单明了,所以整个工作流程既透明又高效。和 BBKNN 一样,
ingest()不会改变数据矩阵本身。与 BBKNN 不同的是,
ingest()能够解决标签映射问题,并且可以保持一个嵌入,这个嵌入可能有我们想要的特性,比如特定的聚类或者轨迹。
我们把这种非对称的数据集整合方式称为从一个标注过的参考数据中摄取注释,然后应用到还没有这个注释的 adata 上。
导入包
import anndata
import scanpy as sc
import pandas as pd
sc.settings.verbosity = 1 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.logging.print_versions()
sc.settings.set_figure_params(dpi=80, frameon=False, figsize=(3, 3), facecolor="white")
PBMCs
我们有一个已经标注好的参考数据集 adata_ref,还有一个想要获取标签和嵌入信息的数据集 adata。
# this is an earlier version of the dataset from the pbmc3k tutorial
adata_ref = sc.datasets.pbmc3k_processed()
adata = sc.datasets.pbmc68k_reduced()
要使用 sc.tl.ingest,这两个数据集必须在相同的变量基础上定义。
var_names = adata_ref.var_names.intersection(adata.var_names)
adata_ref = adata_ref[:, var_names].copy()
adata = adata[:, var_names].copy()
在参考数据集上训练出来的模型和图(这里指的是 PCA以及 UMAP)能够解释参考数据集中观察到的生物学变化。
sc.pp.pca(adata_ref)
sc.pp.neighbors(adata_ref)
sc.tl.umap(adata_ref)
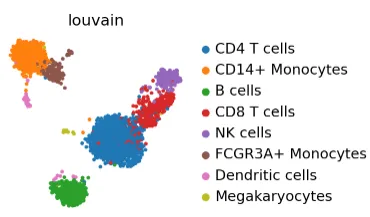
sc.pl.umap(adata_ref, color="louvain")
ingest
我们来根据选定的表示方法,把 adata_ref 里的标签和嵌入信息映射到 adata 里。具体来说,就是用 adata_ref.obsm['X_pca'] 来映射聚类标签和 UMAP 坐标。
sc.tl.ingest(adata, adata_ref, obs="louvain")
adata.uns["louvain_colors"] = adata_ref.uns["louvain_colors"] # fix colors
sc.pl.umap(adata, color=["louvain", "bulk_labels"], wspace=0.5)
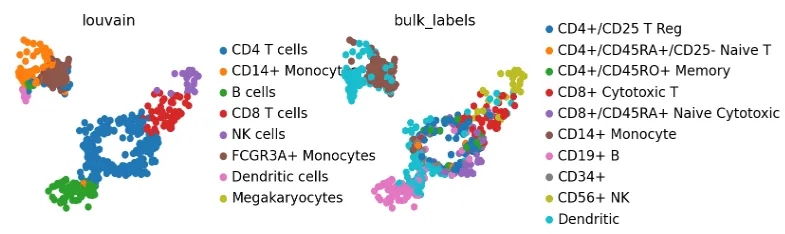
把 “bulk_labels” 注释和 “louvain” 对比一下,可以看出数据整体上是合理地被映射了,不过树突状细胞的注释有点模糊,可能在 adata 里一开始就有这个问题。
adata_concat = anndata.concat([adata_ref, adata], label="batch", keys=["ref", "new"])
adata_concat.obs["louvain"] = (
adata_concat.obs["louvain"]
.astype("category")
.cat.reorder_categories(adata_ref.obs["louvain"].cat.categories)
)
# fix category colors
adata_concat.uns["louvain_colors"] = adata_ref.uns["louvain_colors"]
sc.pl.umap(adata_concat, color=["batch", "louvain"])
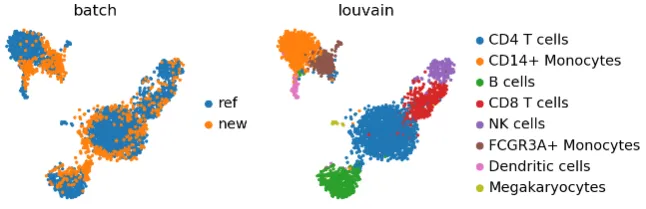
单核细胞和树突状细胞聚类里好像有点批次效应,但新数据在其他地方还算均匀地被映射过去了。
巨核细胞只在 adata_ref 里有,adata 里的细胞都没映射到它上面。要是把参考数据和查询数据换一下,巨核细胞就不会作为一个单独的聚类出现了。这种情况比较极端,因为参考数据量太小了;不过我们得考虑,参考数据里的生物学变化是不是足够丰富,能不能合理地容纳查询数据。
BBKNN
sc.tl.pca(adata_concat)
%%time
sc.external.pp.bbknn(adata_concat, batch_key="batch") # running bbknn 1.3.6
sc.tl.umap(adata_concat)
sc.pl.umap(adata_concat, color=["batch", "louvain"])
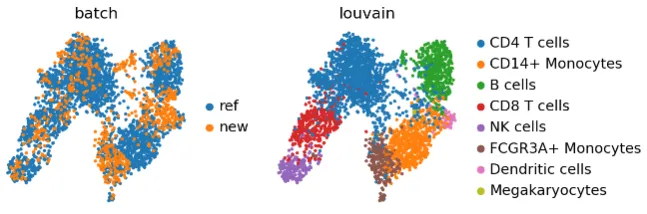
同样,BBKNN 也没有保留巨核细胞群。然而,它似乎使细胞混合得更加均匀。
胰腺
以下数据包含了来自四项不同研究的人类胰腺数据,这些研究在单细胞数据集整合的开创性论文中被使用过,并且此后多次被引用。
# note that this collection of batches is already intersected on the genes
adata_all = sc.read(
"data/pancreas.h5ad",
backup_url="https://www.dropbox.com/s/qj1jlm9w10wmt0u/pancreas.h5ad?dl=1",
)
adata_all.shape # (14693, 2448)
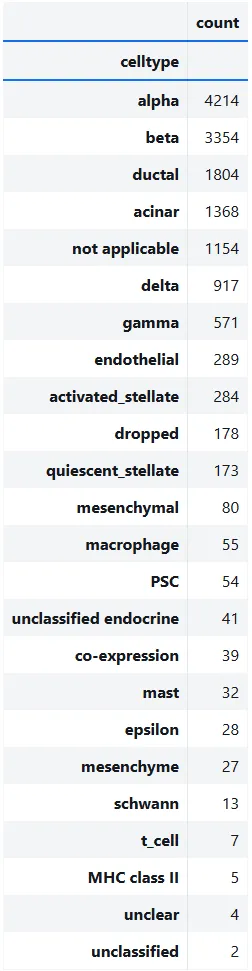
观察这些研究中观察到的细胞类型。
counts = adata_all.obs["celltype"].value_counts()
counts.to_frame()
为了简化可视化,我们移除五个少数类。
minority_classes = counts.index[-5:].tolist() # get the minority classes
# actually subset
adata_all = adata_all[~adata_all.obs["celltype"].isin(minority_classes)].copy()
# reorder according to abundance
adata_all.obs["celltype"] = adata_all.obs["celltype"].cat.reorder_categories(
counts.index[:-5].tolist()
)
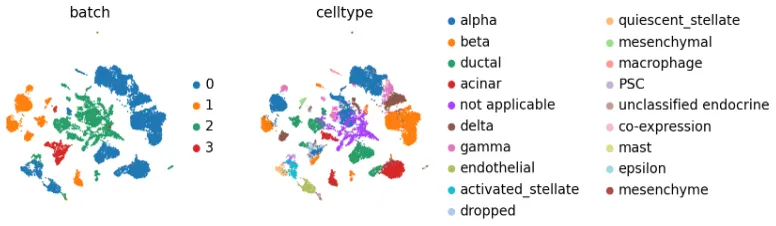
观察批次效应
sc.pp.pca(adata_all)
sc.pp.neighbors(adata_all)
sc.tl.umap(adata_all)
发现批次效应
sc.pl.umap(
adata_all, color=["batch", "celltype"], palette=sc.pl.palettes.vega_20_scanpy
)